Активни съставки: Устекинумаб
STELARA 45 mg инжекционен разтвор
Пакетните вложки Stelara се предлагат за размери на опаковките:- STELARA 45 mg инжекционен разтвор
- STELARA 90 mg инжекционен разтвор
Показания Защо се използва Stelara? За какво е?
Какво е Stelara
Stelara съдържа активното вещество "ustekinumab", моноклонално антитяло.
Моноклоналните антитела са протеини, които разпознават и свързват определени специфични протеини в организма.Стелара принадлежи към група лекарства, наречени "имуносупресори". Тези лекарства намаляват до известна степен активността на имунната система.
За какво е Stelara
Stelara се използва за лечение на следните възпалителни заболявания:
- плакатен псориазис (при възрастни и деца на възраст над 12 години)
- псориатичен артрит (при възрастни)
Плакатен псориазис
Плакатен псориазис е кожно заболяване, което причинява възпаление на кожата и ноктите. Stelara ще намали възпалението и други признаци на заболяването.
Stelara се използва при възрастни с умерен до тежък псориазис на плаки, които не могат да използват циклоспорин, метотрексат или фототерапия или при които тези лечения не работят.
Stelara се използва при деца на възраст от 12 години с умерен до тежък плакатен псориазис, които не могат да понасят фототерапия или други системни терапии, или когато тези лечения не са дали резултат.
Псориатичен артрит
Псориатичният артрит е възпалително заболяване на ставите, обикновено придружено от псориазис. Ако имате активен псориатичен артрит, първо ще бъдете лекувани с други лекарства. Ако не реагирате адекватно на тези лекарства, можете да приемете Stelara за:
- намаляване на признаците и симптомите на заболяването.
- подобряване на физическата функция.
- забавят увреждането на ставите.
Противопоказания Когато Stelara не трябва да се използва
Не използвайте Stelara
- Ако сте алергични към устекинумаб или към някоя от останалите съставки на това лекарство (изброени в точка 6 по -долу)
- Ако имате „активна инфекция, която според Вашия лекар е важна.
Ако не сте сигурни дали някое от горните се отнася за Вас, говорете с Вашия лекар или фармацевт, преди да използвате Stelara.
Предпазни мерки при употреба Какво трябва да знаете, преди да приемете Stelara
Говорете с Вашия лекар или фармацевт, преди да използвате Stelara. Вашият лекар ще проверява вашето здраве преди всяко лечение. Не забравяйте да уведомите Вашия лекар преди всяко лечение за болестите, от които страдате. Кажете също на Вашия лекар, дори ако наскоро сте били в контакт с хора, които може да са имали туберкулоза. Вашият лекар ще Ви прегледа и ще Ви направи изследвания за туберкулоза, преди да Ви даде Stelara. Ако Вашият лекар смята, че сте изложени на риск от туберкулоза, той може да Ви даде лекарства за лечение на туберкулоза.
Пазете се от сериозни странични ефекти
Stelara може да причини сериозни странични ефекти, включително алергични реакции и инфекции. Трябва да обърнете внимание на някои признаци на заболяването, докато приемате Stelara. Вижте "Сериозни нежелани реакции" в точка 4 за пълен списък на тези нежелани реакции.
Преди да използвате Stelara, свържете се с Вашия лекар:
- Ако някога сте имали алергична реакция към Stelara. Попитайте Вашия лекар, ако не сте сигурни.
- Ако някога сте имали някакъв вид рак - това е така, защото имуносупресорите като Stelara частично отслабват имунната система. Това може да увеличи риска от рак.
- Ако имате или сте имали скорошна инфекция.
- Ако някога сте имали нови или променени лезии в областта на псориазис или върху нормална кожа.
- Ако приемате друг вид лечение за псориазис и / или псориатичен артрит - като друг имуносупресор или фототерапия (когато тялото се третира с вид ултравиолетова (UV) светлина). Тези лечения могат също така частично да намалят активността на имунната система. Едновременната употреба на тези терапии със Stelara не е проучена. Възможно е обаче това да увеличи вероятността от заболявания, свързани с отслабване на имунната система.
- Ако използвате или някога сте използвали инжекции за лечение на алергии - не е известно дали Stelara може да ги повлияе.
- Ако сте на 65 или повече години - може да е по -вероятно да получите инфекции
Ако не сте сигурни дали някое от горните състояния се отнася за Вас, говорете с Вашия лекар или фармацевт, преди да започнете лечение със Stelara.
Деца и юноши
Stelara не се препоръчва за лечение на деца (под 12 години), тъй като не е проучен в тази възрастова група.
Взаимодействия Кои лекарства или храни могат да променят ефекта на Stelara
Кажете на Вашия лекар или фармацевт:
- Ако приемате, наскоро сте приемали или е възможно да приемете други лекарства.
- Ако наскоро сте били ваксинирани или Ви предстои ваксинация. Някои видове ваксини (живи ваксини) не трябва да се прилагат, докато използвате Stelara.
Предупреждения Важно е да знаете, че:
Бременност и кърмене
- За предпочитане е да се избягва употребата на Stelara по време на бременност. Ефектите на Stelara върху бременни жени не са известни. Ако сте в детеродна възраст, препоръчително е да избягвате забременяване; трябва да използвате адекватна контрацепция, докато използвате Stelara и поне 15 седмици след спиране на лечението със Stelara. Ако сте бременна, мислите, че може да сте бременна или планирате да забременеете, моля, попитайте Вашия лекар за съвет.
- Ако кърмите или планирате да кърмите, попитайте Вашия лекар за съвет. Вие и Вашият лекар ще решите дали трябва да кърмите или да използвате Stelara. Не може да направи и двете.
Шофиране и работа с машини
Stelara няма или има незначително влияние върху способността за шофиране или работа с машини.
Доза, начин и време на приложение Как да използвате Stelara: Дозировка
Stelara е предназначен за употреба под ръководството и надзора на лекар с опит в диагностиката и лечението на псориазис или псориатичен артрит. Винаги използвайте това лекарство точно както Ви е казал Вашият лекар. Ако се съмнявате, консултирайте се с Вашия лекар. Обсъдете с Вашия лекар кога ще Ви се наложи да си направите инжекции и последващи посещения.
Колко се дава Stelara
Вашият лекар ще реши от колко Stelara се нуждаете и за колко време.
Възрастни от 18 години
- Препоръчителната начална доза е 45 mg Stelara. Пациентите с тегло над 100 килограма (kg) могат да започнат с доза 90 mg вместо 45 mg.
- След първоначалната доза ще вземете следващата доза 4 седмици по -късно, а след това на всеки 12 седмици. Следващите дози обикновено са същите като началната доза.
Деца и юноши на възраст от 12 години
- Вашият лекар ще изчисли правилната доза, включително количеството (обема) на Stelara, което трябва да се инжектира, за да се осигури правилната доза. Правилната доза ще зависи от телесното тегло на детето по време на всяка доза.
- Ако телесното Ви тегло е по -малко от 60 kg, препоръчителната доза е 0,75 mg Stelara на kg телесно тегло.
- Ако телесното тегло е между 60 kg и 100 kg, препоръчителната доза е 45 mg Stelara.
- Ако теглото надвишава 100 kg, препоръчителната доза е 90 mg Stelara.
- След първоначалната доза ще трябва да получите следващата доза след 4 седмици, а след това на всеки 12 седмици.
Как се дава Stelara
- Stelara се прилага като „инжекция под кожата („ подкожно “). В началото на лечението Вашият лекар или медицинска сестра могат да инжектират Stelara.
- Вие и Вашият лекар обаче можете да решите дали можете сами да си инжектирате Stelara. В този случай ще бъдете научени как сами да си инжектирате Stelara.
- За инструкции как да инжектирате Stelara, вижте „Инструкции за приложение“ в края на тази листовка.
Кажете на Вашия лекар, ако имате въпроси относно самостоятелно инжектиране.
Ако сте пропуснали да използвате Stelara
Ако пропуснете доза, свържете се с Вашия лекар или фармацевт. Не вземайте двойна доза, за да компенсирате пропуснатата доза.
Ако сте спрели приема на Stelara
Не е опасно да спрете употребата на Stelara, но ако спрете лечението, вашият псориазис може да се върне.
Ако имате някакви допълнителни въпроси относно употребата на това лекарство, попитайте Вашия лекар или фармацевт.
Предозиране Какво да направите, ако сте приели твърде много Stelara
Ако сте използвали или сте приели твърде много Stelara, незабавно уведомете Вашия лекар или фармацевт. Винаги носете външната кутия на Вашето лекарство със себе си, дори и да е празна.
Странични ефекти Какви са страничните ефекти на Stelara
Както всички лекарства, това лекарство може да предизвика нежелани реакции, въпреки че не всеки ги получава.
Сериозни странични ефекти
Някои пациенти могат да получат сериозни странични ефекти, които може да се нуждаят от спешно лечение.
Алергични реакции - те може да се нуждаят от спешно лечение, затова се свържете с Вашия лекар или потърсете спешна медицинска помощ, ако забележите някой от следните признаци.
- Сериозни алергични реакции ("анафилаксия") са редки при пациенти, приемащи Stelara (засягат до 1 на 1 000 пациенти). Признаците включват:
- затруднено дишане или преглъщане
- ниско кръвно налягане, което може да причини замаяност
- усещане за замаяност или подуване на лицето, устните, устата или гърлото.
- Честите признаци на алергична реакция включват кожен обрив и уртикария (засягат до 1 на 100 души).
Ако имате тежка алергична реакция, Вашият лекар може да реши, че не трябва да използвате отново Stelara.
Инфекции - те може да се нуждаят от спешно лечение, така че незабавно се свържете с Вашия лекар, ако забележите някой от следните признаци.
- Инфекциите на носа и гърлото и обикновената настинка са чести (засягат до 1 на 10 души).
- "Възпалението на подкожната тъкан (" целулит ") е необичайно (засяга до 1 на 100 пациенти).
- Херпес зоостер (вид обрив с мехури) е необичаен (засяга до 1 на 100 пациенти).
Stelara може да намали способността за борба с инфекциите, а някои инфекции могат да станат сериозни.
Трябва да обърнете внимание на признаците на инфекция, докато използвате Stelara. Те включват:
- треска, грипоподобни симптоми, нощно изпотяване
- чувство на умора или недостиг на въздух, упорита кашлица
- гореща, червена, възпалена кожа или болезнен обрив с мехури
- парене при уриниране
- диария
Уведомете незабавно Вашия лекар, ако забележите някой от тези признаци на инфекция. Говорете с Вашия лекар, ако имате някакъв вид инфекция, която продължава или продължава да се връща. Вашият лекар може да реши да спре Stelara, докато инфекцията не се изчисти.Кажете също така на Вашия лекар, ако имате отворени порязвания или рани, които могат да се заразят.
Кожен пилинг - повишеното зачервяване и лющене на кожата върху голяма площ от тялото може да са симптоми на еритродермален псориазис или ексфолиативен дерматит, които са сериозни кожни заболявания. Ако забележите някой от тези признаци, трябва незабавно да уведомите Вашия лекар.
Други странични ефекти
Чести нежелани реакции (засягат до 1 на 10 пациенти):
- Диария
- Гадене
- Чувствам се изморен
- Чувствам се замаян
- Главоболие
- Сърбеж
- Болки в гърба, мускулите или ставите
- Възпалено гърло
- Зъбна инфекция
- Зачервяване и болка на мястото на инжектиране
Нечести нежелани реакции (засягат до 1 на 100 пациенти):
- Депресия
- Течащ или запушен нос
- Кървене, синини, скованост, подуване и сърбеж на мястото на инжектиране
- Увиснал клепач и мускулна релаксация от едната страна на лицето ("парализа на лицето" или "парализа на Бел"), която обикновено е временна
- Промяна в псориазис със зачервяване и нови малки, жълти или бели кожни мехури, понякога придружени от треска (пустулозен псориазис)
- Пилинг на кожата (ексфолиране на кожата)
Редки нежелани реакции (засягат до 1 на 1 000 пациенти)
- Зачервяване и лющене на кожата върху голяма повърхност на тялото, което може да бъде сърбящо или болезнено (ексфолиативен дерматит). Подобни симптоми понякога се развиват като естествена прогресия в типа на симптомите на псориазис (еритродермален псориазис).
Докладване на странични ефекти
Ако получите някакви нежелани реакции, говорете с Вашия лекар или фармацевт.Това включва всички възможни нежелани реакции, които не са изброени в тази листовка. Можете също да съобщите нежелани реакции директно чрез националната система за докладване, изброена в допълнение V. Чрез съобщаване на странични ефекти можете да помогнете за предоставяне на повече информация за безопасността на това лекарство.
Срок на годност и задържане
- Съхранявайте това лекарство на място, недостъпно за деца.
- Съхранявайте в хладилник (2 ° C - 8 ° C). Не замразявайте.
- Съхранявайте флакона във външната опаковка, за да предпазите лекарството от светлина.
- Не разклащайте флаконите на Stelara. Продължителното силно разклащане може да увреди лекарството.
Не използвайте това лекарство
- След срока на годност, отбелязан върху етикета и картонената опаковка след "Годен до:" Срокът на годност се отнася до последния ден от месеца.
- Ако течността е обезцветена, непрозрачна или ако видите плаващи чужди частици (вижте раздел 6 "Описание на това как изглежда Stelara и съдържанието на опаковката").
- Ако знаете или вярвате, че лекарството е било изложено на екстремни температури (например случайно замразено или нагрято).
- Ако продуктът е бил силно разклатен.
- Ако уплътнението е счупено.
Stelara е само за еднократна употреба. Неизползваният продукт, останал във флакона и спринцовката, трябва да се изхвърли.
Не изхвърляйте никакви лекарства през отпадни води или битови отпадъци. Попитайте Вашия фармацевт как да изхвърлите лекарства, които вече не използвате. Това ще помогне за опазването на околната среда.
Краен срок "> Друга информация
Какво съдържа Stelara
- Активната съставка е устекинумаб. Всеки флакон съдържа 45 mg устекинумаб в 0,5 ml
- Другите съставки са: L-хистидин, L-хистидин монохидрохлорид монохидрат, полисорбат 80, захароза, вода за инжекции.
Как изглежда Stelara и какво съдържа опаковката
Stelara е бистър до леко опалесциращ (подобен на перла вид), безцветен до бледожълт инжекционен разтвор.
Разтворът може да съдържа няколко малки полупрозрачни или бели частици протеин. Предлага се в картонена опаковка, съдържаща 1 единична доза, в стъклен флакон от 2 ml.
Всеки флакон съдържа 45 mg устекинумаб в 0,5 ml инжекционен разтвор.
Срок на годност>> Инструкции за администриране
В началото на лечението Вашият лекар ще Ви помогне по време на първата инжекция, но Вие и Вашият лекар можете да решите дали можете сами да си инжектирате Stelara. В този случай ще бъдете научени как сами да инжектирате Stelara. Кажете на Вашия лекар в случай, че ако имате въпроси относно самоинжектирането.
- Не смесвайте Stelara с други инжекционни течности
- Не разклащайте флаконите на Stelara, тъй като енергичното им разклащане може да увреди лекарството. Не използвайте лекарството, ако е било силно разклатено.
Проверете броя на флаконите и подгответе материалите:
Извадете флакона или няколко флакона от хладилника. Оставете флакона извън хладилника за около половин час, което ще позволи на течността да достигне удобна за инжектиране температура (стайна температура).
Провери това:
- броят на флаконите и дозата са правилни
- ако Вашата доза е 45 mg или по -малко, ще вземете 45 mg флакон Stelara
- ако Вашата доза е 90 mg, ще вземете два флакона от 45 mg Stelara и ще трябва да поставите две инжекции. Изберете две различни места по тялото за тези инжекции (например една инжекция в дясното бедро, а другата инжекция в лявото бедро) и продължете с инжекциите една след друга. Използвайте нова игла и нова спринцовка за всяка инжекция.
- лекарството е правилно
- лекарството не е изтекло
- флаконът не е повреден и запушалката е счупена
- разтворът във флакона е бистър или леко опалесциращ (с вид на перла) и безцветен или бледожълт
- течността няма променен или непрозрачен цвят и не съдържа чужди частици
- не е замразен.
Децата с телесно тегло по -малко от 60 kg се нуждаят от доза по -малка от 45 mg. Трябва да сте сигурни в подходящото количество (обем) за отстраняване от флакона и вида на спринцовката, необходима за дозиране. Ако не знаете количеството лекарство или вида на спринцовката, която да използвате, свържете се с Вашия лекар за допълнителни инструкции.
Вземете всичко необходимо и го поставете върху чиста повърхност.Трябва да има спринцовка, игла, антисептични тампони, памучна топка или марля и контейнер за остри предмети.
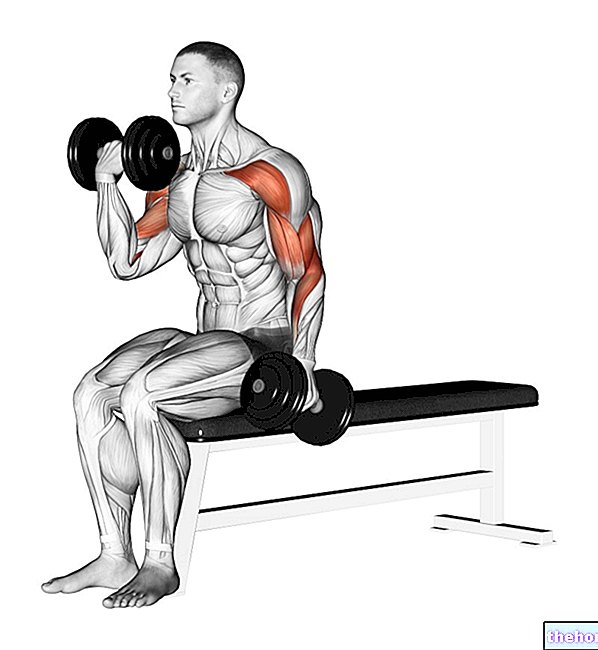
Изберете мястото на инжектиране и го пригответе:
Изберете място за инжектиране.
- Stelara се прилага чрез инжектиране под кожата (подкожно).
- Добро място за инжектиране е горната част на бедрото или около корема (корема) на поне 5 см от пъпа.
- Ако е възможно, не избирайте участъци от кожата с признаци на псориазис.
- Ако някой ви помага по време на инжектирането, той може да избере и горната част на ръцете като място за инжектиране.
Подгответе мястото на инжектиране
- Измийте ръцете си много добре със сапун и топла вода
- Втрийте мястото на инжектиране в кожата с антисептичен тампон
- Не докосвайте отново тази област преди инжектиране.
Пригответе дозата:
- Свалете капачката от горната част на флакона.
- Не сваляйте капачката
- Почистете капачката с антисептичен тампон
- Поставете флакона върху равна повърхност.
- Вземете спринцовката и свалете защитната капачка на иглата.
- Не докосвайте иглата и не оставяйте иглата да докосва нищо.
- Натиснете иглата през гумената запушалка.
- Обърнете флакона и спринцовката с главата надолу.
- Издърпайте буталото на спринцовката, за да напълните спринцовката с количеството течност, както е предписано от Вашия лекар.
- Важно е иглата винаги да е вътре в течността, за да не се образуват въздушни мехурчета в спринцовката.
- Извадете иглата от флакона.
- Задръжте спринцовката с иглата нагоре, за да видите дали вътре няма мехурчета.
- Ако има въздушни мехурчета, леко докоснете отстрани на спринцовката, докато въздушните мехурчета достигнат горната част на спринцовката.
- След това натискайте буталото, докато се отстрани целият въздух (но не и течността). Не оставяйте спринцовката и избягвайте иглата да докосне нещо.
Инжектирайте дозата:
- Внимателно изстискайте частта от чистата кожа, като я държите между палеца и показалеца си. Не стискайте прекалено силно.
- Натиснете иглата в прищипаната кожа.
- Натискайте буталото с палец, докато приключите с инжектирането на цялата течност. Натискайте бавно и стабилно, като държите кожата леко стегната.
- Когато буталото достигне края на спринцовката, издърпайте иглата и освободете кожата.
След инжектирането:
- Натиснете антисептична подложка върху мястото на инжектиране за няколко секунди след инжектирането.
- Възможно е да има малко количество кръв или течност на мястото на инжектиране. Нормално е.
- Можете да натиснете памучна топка или марля върху мястото на инжектиране и да я задържите за 10 секунди.
- Не търкайте кожата на мястото на инжектиране - можете да покриете мястото на инжектиране с малък пластир, ако е необходимо.
Изхвърляне:
- Използваните спринцовки и игли трябва да се поставят в устойчив на пробиване контейнер, например контейнер за остри предмети. За вашето здраве и безопасност и безопасността на другите, никога не използвайте повторно игли или спринцовки. Изхвърлете контейнера за остри предмети в съответствие с местните разпоредби.
- Празните флакони, антисептични кърпички и други устройства могат да се изхвърлят в отпадъците.
Източник на листовката: AIFA (Италианска агенция по лекарствата). Съдържание, публикувано през януари 2016 г. Наличната информация може да не е актуална.
За да имате достъп до най-актуалната версия, препоръчително е да получите достъп до уебсайта на AIFA (Италианска агенция по лекарствата). Отказ от отговорност и полезна информация.
01.0 ИМЕ НА ЛЕКАРСТВЕНИЯ ПРОДУКТ -
СТЕЛАРА РАЗТВОР ЗА ИНЖЕКЦИЯ
02.0 КАЧЕСТВЕН И КОЛИЧЕСТВЕН СЪСТАВ -
STELARA 45 mg инжекционен разтвор
Всеки флакон съдържа 45 mg устекинумаб в 0,5 ml.
STELARA 90 mg инжекционен разтвор
Всеки флакон съдържа 90 mg устекинумаб в 1 ml.
STELARA 45 mg инжекционен разтвор в предварително напълнена спринцовка
Всяка предварително напълнена спринцовка съдържа 45 mg устекинумаб в 0,5 ml.
STELARA 90 mg инжекционен разтвор в предварително напълнена спринцовка
Всяка предварително напълнена спринцовка съдържа 90 mg устекинумаб в 1 ml.
Устекинумаб е напълно човешко, интерлевкиново (IL) -12/23 свързващо IgG1κ моноклонално антитяло, произведено в клетъчна линия на миши миелом, използвайки технология на рекомбинантна ДНК.
За пълния списък на помощните вещества вижте точка 6.1.
03.0 ЛЕКАРСТВЕНА ФОРМА -
STELARA 45 mg инжекционен разтвор
Инжекционен разтвор.
STELARA 90 mg инжекционен разтвор
Инжекционен разтвор.
STELARA 45 mg инжекционен разтвор в предварително напълнена спринцовка
Инжекционен разтвор.
STELARA 90 mg инжекционен разтвор в предварително напълнена спринцовка
Инжекционен разтвор.
Разтворът е бистър до леко опалесциращ, безцветен до бледожълт.
04.0 КЛИНИЧНА ИНФОРМАЦИЯ -
04.1 Терапевтични показания -
Плакатен псориазис
STELARA е показан за лечение на умерен до тежък плакатен псориазис при възрастни пациенти, които не са повлияли, или имат противопоказания или имат непоносимост към други системни терапии, включително циклоспорин, метотрексат (MTX) или PUVA (псорален и ултравиолетови лъчи А) ( вижте точка 5.1).
Плакатен псориазис при педиатрични пациенти
STELARA е показан за лечение на умерен до тежък плакатен псориазис при подрастващи пациенти на възраст над 12 години, които са неадекватно контролирани или непоносими към други системни терапии или фототерапия (вж. Точка 5.1).
Псориатичен артрит (PsA)
STELARA, самостоятелно или в комбинация с MTX, е показан за лечение на активен псориатичен артрит при възрастни пациенти, когато отговорът на предишната терапия с небиологични модифициращи болестта антиревматични лекарства (DMARDs) е неадекватен (вж. Точка 5.1).
болест на Крон
STELARA е показан за лечение на възрастни пациенти с умерена до тежка активна болест на Crohn, които са имали неадекватен отговор, загубили отговор или са установили непоносимост към конвенционалната терапия или антагонист на TNFa или които имат противопоказания за такива терапии.
04.2 Дозировка и начин на приложение -
STELARA трябва да се използва под ръководството и надзора на лекари специалисти с опит в диагностиката и лечението на състоянията, за които е показана STELARA.
Дозировка
Плакатен псориазис
Препоръчителната дозировка на STELARA е начална доза от 45 mg, приложена подкожно, последвана от доза от 45 mg след 4 седмици и на всеки 12 седмици след това.
Трябва да се обмисли прекратяване на лечението при пациенти, които не са показали отговор до 28 седмици лечение.
Пациенти с телесно тегло> 100 кг
При пациенти с тегло над 100 kg началната доза, която трябва да се прилага подкожно, е 90 mg, последвана от доза от 90 mg след 4 седмици и на всеки 12 седмици след това. Дозата от 45 mg също се оказа ефективна при тези пациенти. Дозата от 90 mg обаче показва по -голяма ефикасност (вж. Точка 5.1, таблица 4).
Псориатичен артрит (PsA)
Препоръчителната дозировка на STELARA е начална доза от 45 mg, приложена подкожно, последвана от доза от 45 mg след 4 седмици и на всеки 12 седмици след това. Като алтернатива, 90 mg могат да се използват при пациенти с телесно тегло> 100 kg. Трябва да се обмисли прекратяване на лечението при пациенти, които не са показали отговор до 28 седмици лечение.
Възрастни (≥ 65 години)
Не е необходимо коригиране на дозата при пациенти в напреднала възраст (вж. Точка 4.4).
Бъбречна и чернодробна недостатъчност
STELARA не е проучен при тази популация пациенти. Не може да се направи препоръка относно дозата, която трябва да се даде.
Педиатрична популация
Безопасността и ефикасността на STELARA при деца с псориазис под 12 години или при деца с псориатичен артрит под 18 години все още не са установени.
Плакатен псориазис при педиатрични пациенти (на възраст от 12 години)
Препоръчителната доза STELARA въз основа на телесното тегло е показана в таблиците по -долу (Таблици 1 и 2). STELARA трябва да се дава на седмици 0 и 4, а след това на всеки 12 седмици.
Таблица 1: Препоръчителна доза STELARA за педиатрични пациенти с псориазис
a За изчисляване на обема на инжектиране (mL) за телесното тегло на пациента (kg) x 0,0083 (ml / kg) или вижте Таблица 2. Изчисленият обем трябва да бъде закръглен до най -близките 0,01 ml и да се приложи с помощта на градуирана спринцовка от 1 ml. .
Таблица 2: Обеми на инжектиране на STELARA за педиатрични пациенти
Трябва да се обмисли прекратяване на лечението при пациенти, които не показват отговор до 28 седмици лечение.
болест на Крон
В схемата на лечение първата доза STELARA се прилага интравенозно. За дозировката на режима на интравенозно дозиране, вижте точка 4.2 от КХП на STELARA 130 mg концентрат за инфузионен разтвор.
Първото подкожно приложение на 90 mg STELARA трябва да се извърши на 8 -та седмица след интравенозната доза. След това се препоръчва дозиране на всеки 12 седмици.
Пациентите, които не са показали адекватен отговор 8 седмици след първата подкожна доза, могат след това да получат втора подкожна доза (вж. Точка 5.1).
Пациентите, които не са успели да дозират на всеки 12 седмици, могат да се възползват от увеличаване на честотата на дозиране на всеки 8 седмици (вж. Точка 5.1).
Пациентите могат да получават дозата на всеки 8 седмици или на всеки 12 седмици след това въз основа на клинична преценка (вж. Точка 5.1).
Трябва да се обмисли прекратяване на лечението при пациенти, които не показват доказателства за терапевтична полза на 16 -та или 16 -та седмица след преминаване към дозиране на всеки 8 седмици.
Имуномодулаторите и / или кортикостероидите могат да продължат по време на лечението със STELARA. При пациенти, които са се повлияли от лечението със STELARA, кортикостероидите могат да бъдат намалени или отменени в съответствие със стандартните грижи.
Ако терапията се прекъсне, възобновяването на лечението с подкожно приложение на всеки 8 седмици е безопасно и ефективно.
Възрастни (≥ 65 години)
Не е необходимо коригиране на дозата при пациенти в напреднала възраст (вж. Точка 4.4).
Бъбречна и чернодробна недостатъчност
STELARA не е проучен при тази популация пациенти. Не може да се направи препоръка относно дозата, която трябва да се даде.
Педиатрична популация
Безопасността и ефикасността на STELARA при лечението на болестта на Crohn при деца под 18 години все още не са установени. Няма налични данни.
Начин на приложение
STELARA 45 mg и 90 mg във флакони или предварително напълнени спринцовки е формулирана само за подкожно инжектиране. Ако е възможно, избягвайте инжектирането в области, засегнати от псориазис.
След като получат адекватни инструкции относно техниката на подкожно инжектиране, пациентите или техните полагащи грижи могат да прилагат STELARA, ако лекарят прецени за необходимо. Лекарят обаче трябва да осигури подходящо периодично наблюдение на пациентите. Пациентите или болногледачите трябва да бъдат инструктирани да прилагат предписаното количество STELARA, както е указано в листовката. Пълни инструкции за приложение са дадени в листовката.
За допълнителна информация относно подготовката и специални предпазни мерки при работа, вижте точка 6.6.
04.3 Противопоказания -
Свръхчувствителност към активното вещество или към някое от помощните вещества, изброени в точка 6.1.
Активна, клинично значима инфекция (напр. Активна туберкулоза; вж. Точка 4.4).
04.4 Специални предупреждения и подходящи предпазни мерки при употреба -
Инфекции
Устекинумаб може да увеличи риска от получаване на инфекции и реактивиране на латентни.
В някои клинични проучвания са наблюдавани сериозни бактериални, гъбични и вирусни инфекции при пациенти, получаващи STELARA (вж. Точка 4.8).
Трябва да се внимава, когато се обмисля употребата на STELARA при пациенти с хронична инфекция или с анамнеза за повтаряща се инфекция (вж. Точка 4.3).
Преди започване на лечение със STELARA, всички пациенти трябва да бъдат оценени за наличие на туберкулозна инфекция. STELARA не трябва да се прилага при пациенти с активна туберкулоза (вж. Точка 4.3). Лечението на латентна туберкулозна инфекция трябва да се започне преди прилагане на STELARA.Туберкулозната терапия трябва да се обмисли преди започване на STELARA при пациенти с анамнеза за латентна или активна туберкулоза, които не са Адекватна терапевтична пътека може да бъде потвърдена Пациентите на терапия със STELARA трябва да бъдат внимателно наблюдавани за признаци и симптоми на активна туберкулоза, по време и след лечението.
Пациентите трябва да бъдат посъветвани да потърсят лекарска помощ, ако забележат признаци и симптоми, които могат да показват „продължаваща инфекция. Ако пациентът развие тежка“ инфекция, те трябва да бъдат наблюдавани внимателно и STELARA не трябва да се прилага, докато „инфекцията не изчезне.
Новообразувания
Имуносупресорите като устекинумаб могат да увеличат риска от развитие на рак.
Някои пациенти, които са получавали STELARA в клинични изпитвания, развиват кожни и не-кожни злокачествени заболявания (вж. Точка 4.8).
Не са провеждани клинични проучвания, включващи пациенти с анамнеза за злокачествено заболяване или при които лечението със STELARA е продължило въпреки появата на продължаващи злокачествени заболявания. Ето защо трябва да се внимава, когато се обмисля лечение със STELARA при тези пациенти.
Всички пациенти, особено тези на възраст над 60 години, пациенти с анамнеза за продължителна имуносупресивна терапия или с анамнеза за лечение с PUVA, трябва да бъдат наблюдавани за наличие на немеланомен рак на кожата (вж. Точка 4.8).
Реакции на свръхчувствителност
В постмаркетинговия опит са докладвани сериозни реакции на свръхчувствителност, в някои случаи дори няколко дни след лечението.Настъпили са анафилаксия и ангиоедем.
Чувствителност към латекс
Капачката на иглата на предварително напълнената спринцовка STELARA е изработена от сух естествен каучук (производно на латекс), който може да предизвика алергични реакции при чувствителни към латекс индивиди.
Ваксинации
Препоръчва се да не се прилагат живи вирусни или бактериални ваксини (като Calcilte и Guérin bacillus, BCG) едновременно с лечението със STELARA. Не са провеждани специфични клинични проучвания при пациенти, на които наскоро са поставени живи вирусни или бактериални ваксини. Няма данни за вторично предаване на инфекции с жива ваксина при пациенти, получаващи STELARA. Преди прилагане на жива вирусна или бактериална ваксина, лечението със STELARA трябва да бъде спряно поне 15 седмици след последното приложение и може да бъде възобновено не по -рано от 2 седмици след ваксинацията. ваксина, за да се възползвате от допълнителни данни и насоки за едновременната употреба на имуносупресори след ваксинация.
Пациентите на терапия със STELARA могат да бъдат лекувани едновременно с инактивирани или неживи ваксини.
Продължителното лечение със STELARA не потиска хуморалния имунен отговор към пневмококов полизахарид или ваксина срещу тетанус (вж. Точка 5.1).
Съпътстваща имуносупресивна терапия
Безопасността и ефикасността на STELARA в комбинация с други имуносупресори, включително биологични агенти или фототерапия, не са оценявани в проучвания за псориазис. В проучвания за болестта на Crohn, едновременната употреба на имуносупресори или кортикостероиди не изглежда да повлияе на безопасността или ефикасността на STELARA.
Трябва да се внимава, когато се обмисля едновременната употреба на други имуносупресори и STELARA, или когато е резултат от лечение с други биологични имуносупресори (вж. Точка 4.5).
Имунотерапия
STELARA не е оценяван при пациенти, подложени на алергична имунотерапия.
Не е известно дали STELARA може да повлияе алергичната имунотерапия.
Тежки кожни състояния
При пациенти с псориазис се съобщава за ексфолиативен дерматит след лечение с устекинумаб (вж. Точка 4.8). Пациентите с плакатен псориазис могат да развият еритродермален псориазис със симптоми, които могат да бъдат клинично неразличими от ексфолиативен дерматит, като естествен ход на заболяването. Като част от наблюдението на пациенти с псориазис, лекарите трябва да обърнат внимание на симптомите на еритродермален псориазис или ексфолиативен дерматит. Ако се появят тези симптоми, трябва да се започне подходяща терапия. STELARA трябва да се преустанови, ако се подозира лекарствена реакция.
Специални популации
Възрастни (≥ 65 години)
Като цяло не са наблюдавани разлики в ефикасността или безопасността на STELARA при пациенти на възраст 65 или повече години в сравнение с по -младите пациенти, но броят на пациентите на възраст 65 или повече години не е достатъчен, за да се определи дали те реагират по различен начин, отколкото при по -младите пациенти. по -висока честота на инфекции сред възрастното население като цяло, трябва да се внимава при лечение на пациенти в напреднала възраст.
04.5 Взаимодействия с други лекарствени продукти и други форми на взаимодействие -
Живите ваксини не трябва да се прилагат едновременно със STELARA (вж. Точка 4.4).
Не са провеждани проучвания за взаимодействия при хора. В популационните фармакокинетични анализи на проучванията фаза III е изследван ефектът от най -често използваните съпътстващи лекарства при пациенти с псориазис (включително парацетамол, ибупрофен, ацетилсалицилова киселина)., Метформин, аторвастатин, левотироксин ) върху фармакокинетичния профил на устекинумаб. Не е установено взаимодействие с тези едновременно прилагани лекарствени продукти. Основата за този анализ е наличието на най -малко 100 пациенти (> 5% от изследваната популация), лекувани едновременно с тези лекарствени продукти в продължение на поне 90% от периода на изследването. При пациенти с псориатичен артрит или болест на Crohn, фармакокинетиката на устекинумаб не се повлиява от едновременната употреба на МТХ, НСПВС, 6-меркаптопурин, азатиоприн и перорални кортикостероиди, или от предишно излагане на анти-TNFα агенти. инвитро не показват необходимостта от коригиране на дозата при пациенти, приемащи едновременно субстрати на CYP450 (вж. точка 5.2).
В проучвания за псориазис профилите на безопасност и ефикасност на STELARA, прилаган в комбинация с имуносупресори, включително биологични агенти или фототерапия, не са оценени. В проучвания за псориатичен артрит не изглежда, че едновременната употреба на МТХ повлиява безопасността и ефикасността на STELARA. В проучванията на болестта на Crohn едновременната употреба на имуносупресори или кортикостероиди не изглежда да повлиява безопасността или ефикасността на STELARA. (Вж. Точка 4.4).
04.6 Бременност и кърмене -
Жени с детероден потенциал
Жените с детероден потенциал трябва да използват ефективни методи за контрацепция по време на лечението и най -малко 15 седмици след прекратяване на лечението.
Бременност
Няма достатъчно данни за употребата на устекинумаб по време на бременност. Изследванията при животни не показват преки или косвени вредни ефекти по отношение на бременността, ембрионалното / феталното развитие, раждането или постнаталното развитие (вж. Точка 5.3). е за предпочитане да се избягва употребата на STELARA по време на бременност.
Време за хранене
Не е известно дали устекинумаб се екскретира в кърмата. Някои клинични проучвания при животни показват екскрецията на ниски нива на устекинумаб в кърмата.Не е известно дали устекинумаб се абсорбира системно след поглъщане. Като се има предвид способността на устекинумаб да предизвиква нежелани реакции при кърмачета, решението дали да се преустанови кърменето по време на лечението и до 15 седмици след прекратяване, или прилагането на терапия със STELARA трябва да се вземе предвид ползата от лечението. " бебето и ползата от лечението със STELARA за майката.
Плодовитост
Ефектите на устекинумаб върху фертилитета при хора не са оценени (вж. Точка 5.3).
04.7 Ефекти върху способността за шофиране и работа с машини -
STELARA няма или има незначително влияние върху способността за шофиране или работа с машини.
04.8 Нежелани реакции -
Обобщение на профила на безопасност
Най -честите нежелани реакции с устекинумаб (> 5%) в контролираните фази на псориазис, псориатичен артрит и болестта на Crohn клиничните изпитвания при възрастни са назофарингит и главоболие. нежеланите реакции, съобщени при STELARA, са тежки реакции на свръхчувствителност, включително анафилаксия (вж. точка 4.4). Общият профил на безопасност е сходен при пациенти с псориазис, псориатичен артрит и болест на Crohn.
Обобщена таблица на нежеланите реакции
Данните за безопасност, отчетени по -долу, отразяват експозицията на устекинумаб при възрастни в 12 клинични проучвания фаза II и фаза III, включващи 5 884 пациенти (4 135 с псориазис и / или псориатичен артрит и 1749 с болест на Crohn). Това включва експозиция на STELARA в контролирани и неконтролирани фази на клинични изпитвания за най -малко 6 месеца или 1 година (съответно 4 105 и 2846 пациенти с псориазис, псориатичен артрит или болест на Crohn) с експозиция в продължение на най -малко 4 или 5 години (съответно 1482 и 838 пациенти) с псориазис).
Таблица 3 представя списък на нежеланите реакции от клинични проучвания при псориазис, псориатичен артрит и болестта на Crohn при възрастни, както и нежелани реакции, съобщени от постмаркетинговия опит. Нежеланите лекарствени реакции са изброени по системо -органни класове и честота, като се използва следната конвенция: Много чести (≥ 1/10), Чести (≥ 1/100 до
В рамките на всеки честотен клас нежеланите реакции се съобщават в низходящ ред по тежест.
Таблица 3 - Списък на нежеланите реакции
Описание на избрани нежелани реакции
Инфекции
В някои плацебо-контролирани проучвания при пациенти с псориазис, псориатичен артрит и болест на Crohn, честотата на инфекция или тежка инфекция е сходна между пациентите, лекувани с ustekinumab и тези, лекувани с плацебо. Във фазата на плацебо лечение в клинични изпитвания при пациенти с псориазис, пациенти с псориатичен артрит и пациенти с болест на Crohn, честотата на инфекцията е 1,38 на пациент-година на проследяване при пациенти, получаващи устекинумаб и 1,35 при тези, получаващи плацебо. Случаи на тежки инфекции са настъпили до 0,03 на пациент-година от проследяване при пациенти, лекувани с устекинумаб (27 сериозни инфекции за 829 пациент-години на проследяване) и 0,03 при пациенти, лекувани с плацебо (11 тежки инфекции за 385 пациент-години на проследяване) (вижте точка 4.4).
В контролирани и неконтролирани фази на клинични изпитвания при псориазис, псориатичен артрит и болест на Crohn, представляващи 10 953 пациент-години експозиция при 5884 пациенти, проследяване средната стойност е 0,99 години; 3,2 години за проучвания за псориазис, 1,0 година за проучвания за псориатичен артрит и 0,6 години за проучвания за болестта на Crohn.Честотата на инфекцията е 0,91 на пациент-година на проследяване при пациенти, лекувани с устекинумаб, и честотата на сериозните инфекции е била 0,02 на пациент-година от проследяване при пациенти, получаващи устекинумаб (178 сериозни инфекции за 10 953 пациент-години на проследяване) и докладвани сериозни инфекции, включително анален абсцес, целулит, пневмония, дивертикулит, гастроентерит и вирусни инфекции.
В клинични изпитвания пациентите с латентна туберкулоза, лекувани едновременно с изониазид, не развиват туберкулоза.
Новообразувания
В плацебо-контролираните фази на клинични изпитвания при псориазис, псориатичен артрит и болест на Crohn, честотата на злокачествени заболявания, с изключение на немеланомен рак на кожата, е 0,12 на 100 пациент-години на проследяване за пациенти, лекувани с устекинумаб (1 пациент от 829 пациент-години на проследяване) в сравнение с 0,26 за пациенти, лекувани с плацебо (1 пациент от 385 пациент-години на проследяване). Честотата на немеланомен рак на кожата е 0,48 на 100 пациент-години проследяване за пациенти на терапия с устекинумаб (4 пациенти от 829 пациент-години на проследяване) в сравнение с 0,52 за пациенти, лекувани с плацебо (2 пациенти от 385 пациент-години на проследяване).
В контролирани и неконтролирани фази на клинични изпитвания при псориазис, псориатичен артрит и болест на Crohn, представляващи 10 935 пациент-години експозиция при 5 884 пациенти, проследяване медианата е 1,0 година; 3,2 години за изследванията на псориазис, 1,0 години за проучванията за псориатичен артрит и 0,6 години за изследванията на болестта на Crohn. проследяване (честота от 0,53 на 100 пациент-години от проследяване за пациенти, лекувани с устекинумаб). Честотата на злокачествени заболявания, съобщени при пациенти, лекувани с устекинумаб, е сравнима с очакваната честота в общата популация (стандартизиран процент на заболеваемост = 0,87 [95% доверителен интервал: 0,66, 1,14], коригиран според възрастта, пола и расата). Най-често наблюдаваните злокачествени заболявания, различни от немеланомен рак на кожата, са рак на простатата, меланом, колоректален рак и рак на гърдата. Честотата на немеланомен рак на кожата е 0,49 на 100 пациент-години от проследяване за пациенти, лекувани с устекинумаб (53 пациенти от 10 919 пациент-години на проследяване). Съотношението на пациентите с базално -клетъчен рак на плоскоклетъчен рак на кожата (4: 1) е сравнимо с очакваното съотношение в общата популация (вж. Точка 4.4).
Реакции на свръхчувствителност
По време на контролираните фази на псориазис и псориатичен артрит клиничните изпитвания на устекинумаб, обрив и уртикария са наблюдавани при
Имуногенност
В клинични изпитвания при псориазис и псориатичен артрит по -малко от 8% от пациентите, приемащи устекинумаб, развиват антитела към устекинумаб. В клинични изпитвания при болестта на Crohn, по-малко от 3% от лекуваните с устекинумаб пациенти развиват антитела към устекинумаб. Не се наблюдава очевидна връзка между развитието на антитела към устекинумаб и развитието на реакциите на мястото на инжектиране.Повечето пациенти, положителни за антитела срещу антиустекинумаб, са имали неутрализиращи антитела.Ефикасността на лечението е била по-ниска при положителни пациенти. положителността на антителата обаче не изключва клиничен отговор.
Педиатрична популация
Нежелани реакции при педиатрични пациенти от 12 -годишна възраст с плакатен псориазис
Безопасността на устекинумаб е проучена в проучване фаза 3, включващо 110 пациенти на възраст 12-17 години за период до 60 седмици. Нежеланите събития, съобщени в това проучване, са подобни на тези, наблюдавани в предишни проучвания при възрастни с псориазис на плаки.
Докладване на предполагаеми нежелани реакции
Съобщаването на предполагаеми нежелани реакции, настъпили след разрешаване на лекарствения продукт, е важно, тъй като позволява непрекъснато проследяване на съотношението полза / риск на лекарствения продукт.
04.9 Предозиране -
Еднократни дози от лекарствения продукт до 6 mg / kg са прилагани интравенозно в клинични проучвания, без да се наблюдава появата на ограничаваща дозата токсичност. В случай на предозиране се препоръчва пациентът да бъде наблюдаван за признаци или симптоми на нежелани реакции и незабавно да се започне подходяща симптоматична терапия.
05.0 ФАРМАКОЛОГИЧНИ СВОЙСТВА -
05.1 "Фармакодинамични свойства -
Фармакотерапевтична група: Имуносупресори, интерлевкинови инхибитори, ATC код: L04AC05.
Механизъм на действие
Устекинумаб е изцяло човешко IgG1κ моноклонално антитяло, което специфично свързва р40 протеина, споделена субединица на интерлукин (IL) -12 и IL -23, човешки цитокини. Устекинумаб инхибира биологичната активност на човешки IL-12 и IL-23, като предотвратява свързването на р40 с рецепторния протеин на IL-12Rb1, експресиран на повърхността на имунните клетки. Устекинумаб не може да се свърже с IL-12 или IL-23, които вече са свързани към IL-12Rb1 рецептори, присъстващи на клетъчната повърхност.Така че е малко вероятно устекинумаб да допринесе за комплемент-медиирана или антитяло-медиирана цитотоксичност на клетки с IL-12 и / или IL-23 рецептори. IL-12 и IL-23 са хетеродимерни цитокини, секретирани от активирани антиген-представящи клетки, като макрофаги и дендритни клетки, и двата цитокина участват в имунната активност; IL-12 стимулира клетките естествен убиец (NK) и води до диференциация на CD4 + Т клетки към Т фенотип помощник 1 (Th1), IL-23 индуцира пътека на Т. помощник 17 (Th17). Ненормалното регулиране на IL-12 и IL-23 е свързано с имунно-медиирани заболявания, като псориазис, псориатичен артрит и болест на Crohn.
Чрез свързване към споделената р40 субединица на IL-12 и IL-23, устекинумаб може да прояви клиничните си ефекти при псориазис, псориатичен артрит и болест на Crohn, като наруши цитокиновите пътища Th1 и Th17, които са от решаващо значение за заболяването. На тези заболявания. При пациенти с болест на Crohn лечението с устекинумаб води до намаляване на възпалителните индекси, включително С-реактивен протеин (CRP) и фекален калпротектин по време на фазата на индукция; тази индукция след това се поддържа през поддържащата фаза.
Имунизация
По време на дългосрочното удължаване на Проучване 2 за псориазис (PHOENIX 2), възрастни пациенти, лекувани със STELARA в продължение на най-малко 3,5 години, показват подобни реакции на антитела както към пневмококова полизахаридна, така и срещу тетанусна ваксина като група за контрол на псориатични пациенти, лекувани с несистемни лекарства .Подобна част от възрастните пациенти развиват защитни нива на антипневмококови и антитетанусни антитела и титрите на антителата са сходни между пациентите, лекувани със STELARA, и пациентите в контролната група.
Клинична ефикасност и безопасност
Плакатен псориазис (възрастни)
Профилите на ефикасност и безопасност на устекинумаб са оценени при 1 996 пациенти в две рандомизирани, двойно-слепи, плацебо-контролирани клинични проучвания, проведени при пациенти с умерен до тежък плакатен псориазис, които са кандидати за фототерапия или за системна терапия. В допълнение, активно, контролирано от лечение, рандомизирано, заслепено от оценители клинично изпитване сравнява устекинумаб и етанерцепт при пациенти с умерен до тежък псориазис с плаки, които реагират неадекватно или имат непоносимост или имат противопоказания за циклоспорин, MTX или PUVA.
Изследване на псориазис 1 (PHOENIX 1) оценява 766 пациенти. От тях 53% не са повлияли, не са имали непоносимост или са имали противопоказания за друга системна терапия.Пациентите, произволно назначени на устекинумаб, са лекувани с дози от 45 mg или 90 mg на 0 и 4 седмици и впоследствие със същата доза на всеки 12 седмици. , които бяха рандомизирани в групата на плацебо лечение на 0 и 4 седмици, преминаха на устекинумаб (45 mg или 90 mg) на 12 и 16 седмици, последвано от една доза на всеки 12 седмици.Пациенти, първоначално рандомизирани на ustekinumab, които постигнаха отговор на 75 в индекса Индекс на площ и тежест на псориазис (PASI) (подобрение на PASI най-малко 75% спрямо изходното ниво) на 28 и 40 седмици, бяха повторно рандомизирани и назначени в групата на лечение с ustekinumab, прилагана на всеки 12 седмици, или в групата на плацебо (т.е. спиране на терапията) . Пациентите, повторно рандомизирани в плацебо групата на 40-та седмица, възобновиха приема на устекинумаб с първоначалния си график на дозиране, ако са загубили поне 50% от подобрението на PASI, постигнато на 40-та седмица. Всички пациенти са проследени общо 76 седмици след първото приложение на лекарството за изследване.
Проучване 2 на псориазис (PHOENIX 2) оценява 1230 пациенти. От тях 61% не реагират, имат непоносимост или имат противопоказания за „друга системна терапия. Пациентите, произволно назначени на устекинумаб, са лекувани с дози от 45 mg или 90 mg на седмици 0 и 4, а след това с допълнителна доза на седмица 16. бяха рандомизирани в групата на плацебо лечение на седмици 0 и 4, преминаха на устекинумаб (45 mg или 90 mg) на седмици 12 и 16. Всички пациенти бяха проследени общо 52 седмици след първото приложение на изследваното лечение.
Проучване 3 на псориазис (ACCEPT) оценява 903 пациенти с умерен до тежък псориазис, които се повлияват неадекватно или имат непоносимост или имат противопоказания за други системни терапии, сравнявайки ефикасността на устекинумаб спрямо етанерцепт и оценявайки безопасността на двата биопрепарата при пациенти. 12-седмичен период на активен контрол на проучването, пациентите бяха рандомизирани да получават етанерцепт (50 mg два пъти седмично), устекинумаб 45 mg на 0 и 4 седмици или 90 mg ustekinumab на 0 и 4 седмица.
В клинични изпитвания 1 и 2 при псориазис, изходните характеристики на заболяването обикновено се припокриват във всички лечебни групи със среден изходен PASI резултат, вариращ от 17 до 18, „псориатична област на телесната повърхност (Площ на повърхността на тялото, BSA) медиана ≥ 20 и среден дерматологичен индекс на качеството на живот (Дерматологичен индекс за качество на живот, DLQI) между 10 и 12. Около една трета (изследване на псориазис 1) и една четвърт (изследване на псориазис 2) от пациентите са имали псориатичен артрит (PsA). Подобна тежест на заболяването е наблюдавана и в Проучване 3 на псориазис.
L "крайна точка Основен в тези проучвания е делът на пациентите, които са постигнали отговор на PASI 75 от изходното ниво на седмица 12 (вж. Таблици 4 и 5).
Таблица 4 - Обобщение на клиничния отговор в проучване 1 за псориазис (PHOENIX 1) и проучване 2 (PHOENIX 2)
на стр
b PGA = (Глобална оценка на лекаря) глобална оценка на лекаря
Таблица 5 - Обобщение на клиничния отговор на седмица 12 в проучване 3 за псориазис (ACCEPT)
на стр
b р = 0,012 за устекинумаб 45 mg спрямо етанерцепт.
В проучване 1 за псориазис, поддържането на оценка PASI 75 е значително по -високо при продължаване на лечението, отколкото при преустановяване на лечението (p
При пациенти, повторно рандомизирани на плацебо, които са възобновили приема на устекинумаб в първоначалния им режим след ≥ 50% загуба на подобрение на PASI, 85% са възстановили PASI 75 отговор в рамките на 12 седмици след повторното въвеждане на терапията. В проучване 1 за псориазис, на 2 -ра и 12 -та седмица, са наблюдавани значителни подобрения в изходния DLQI във всяка група на лечение с ustekinumab в сравнение с групата на плацебо. Подобрението се поддържа до седмица 28. По подобен начин се наблюдават значителни подобрения в Проучване 2 на псориазис на 4 и 12 седмици, които се запазват до 24 седмица. В проучване 1 на псориазис подобренията при псориазис също са значителни. Индекс на тежест на псориазис на ноктите), общите резултати от умствения и физическия компонент на SF-36 и визуалната аналогова скала (Визуална аналогова скала, VAS) за сърбеж във всяка група на лечение с ustekinumab в сравнение с плацебо. В проучване 2 за псориазис, скалата HADS (Болнична скала за тревожност и депресия) и въпросника на WLQ (Въпросник за работни ограничения) във всяка терапевтична група с устекинумаб спрямо плацебо.
Псориатичен артрит (PsA) (възрастни)
Доказано е, че устекинумаб подобрява признаците и симптомите, физическата функция и качеството на живот, свързани със здравето, и намалява скоростта на прогресия на периферните ставни увреждания при възрастни пациенти с активен PsA.
Безопасността и ефикасността на устекинумаб са оценени при 927 пациенти в две рандомизирани, двойно-слепи, плацебо-контролирани клинични изпитвания при пациенти с активен PsA (≥ 5 подути и ≥ 5 болезнени стави) въпреки нестероидната противовъзпалителна терапия (НСПВС). ) или модифицираща болестта антиревматична медикаментозна терапия (DMARD). Пациентите в тези проучвания са били диагностицирани с PsA в продължение на най-малко 6 месеца. Пациентите с всеки подтип на PSA са били записани, включително полиартикуларен артрит без данни за ремавтоиди на възли (39%), спондилит с периферен артрит (28%), периферен асиметричен артрит (21%), засягане на дисталните междуфалангови стави (12%) и осакатяващ артрит (0,5%). Над 70%и 40%от пациентите в двете проучвания са имали ентезит и дактилит при Пациентите са рандомизирани да получават устекинумаб 45 mg, 90 mg или плацебо подкожно на седмици 0 и 4, последвано от a
приложение на всеки 12 седмици (q12w). Приблизително 50% от пациентите продължават със стабилни дози MTX (≤ 25 mg / седмица).
В PsA проучване 1 (PSUMMIT I) и PsA проучване 2 (PSUMMIT II) съответно 80% и 86% от пациентите са били лекувани с DMARDs. Предишно лечение с антитуморни некротични фактори (TNF) α агенти не беше позволено в проучване 1. В проучване 2 по-голямата част от пациентите (58%, n = 180) преди това са получавали едно или повече лечения с анти-TNFα агент, от които повече от 70% са спрели лечението с анти-TNFα по всяко време за загуба на ефикасност или нетърпимост.
Знаци и симптоми
Лечението с устекинумаб доведе до значителни подобрения в оценката на активността на заболяването в сравнение с плацебо на 24 -та седмица. Първичната крайна точка беше процентът на пациентите, постигнали отговор на Американския колеж по ревматология (ACR) 20 на 24 -та седмица. I Ключовите резултати за ефикасност са показани в следващата таблица 6 . Таблица 6 - Брой пациенти, постигнали клиничен отговор в проучване 1 за псориатичен артрит (PSUMMIT I) и проучване 2 (PSUMMIT II) на 24 -та седмица
на стр
b стр
c p = NS
d Брой пациенти с увреждане на псориазис на кожата при изходно ниво BSA ≥ 3%
Отговорите ACR 20, 50 и 70 непрекъснато се подобряват или остават постоянни през седмица 52 (PsA проучване 1 и 2) и седмица 100 (PsA проучване 1). В проучване 1 на PsA отговорите на ACR 20 на седмица 100 са постигнати съответно с 57% и 64% за 45 mg и 90 mg. В проучване 2 на PsA отговорите на ACR 20 на 52 -та седмица са постигнати съответно с 47% и 48% за 45 mg и 90 mg.
Процентът на пациентите, постигнали отговор по критериите за реакция на модифициран псориатичен артрит (PsARC), също е значително по-висок в групата с ustekinumab в сравнение с плацебо на 24-та седмица. Отговорите на PsARC се поддържат през 52 и 100 седмици. лекуваните пациенти, които са имали спондилит с периферен артрит като първоначална проява, показват 50 и 70 процентно подобрение в индекса на активност на болестта на анкилозиращия спондилит на банята (BASDAI) в сравнение с плацебо на 24 -та седмица. тези, които не получават МТХ и се поддържат през 52 и 100 седмици. Пациентите, лекувани преди това с анти-TNFα агенти, които са получавали устекинумаб, постигат по-голям отговор на 24-та седмица в сравнение с пациентите, които са получавали плацеб или (отговорът на ACR 20 на седмица 24 за 45 mg и 90 mg е съответно 37%и 34%, в сравнение с 15%плацебо; стр
При пациенти с ентезит и / или дактилит на изходно ниво е наблюдавано значително подобрение на оценката на ентезит и дактилит в групата с устекинумаб в сравнение с групата на плацебо в седмица 24 в проучване на PsA 2. Значително подобрение в оценката на ентезита и числено (не статистически значимо ) подобрение на резултата от дактилит в групата с 90 mg устекинумаб (р = NS) в сравнение с плацебо на 24 -та седмица. Подобренията в резултата за ентезит и дактилит се поддържат през 52 и 100 седмици.
Рентгенографски отговор
Структурните увреждания както в ръцете, така и в краката се изразяват като промяна в общия резултат на ван дер Хайде-Шарп (vdH-S резултат), модифициран за PsA чрез добавяне на дистални междуфалангови стави на ръката, от изходното ниво. Извършен е предварително специфичен интегриран анализ комбиниране на данни от 927 субекта както от PsA проучване 1, така и от проучване 2.
Устекинумаб демонстрира статистически значимо намаляване на скоростта на прогресиране на структурните увреждания в сравнение с плацебо, измерено чрез промяната от изходното ниво до седмица 24 в модифицирания общ резултат на vdH-S (среден ± SD резултат е 0,97 ± 3,85 в групата на плацебо спрямо 0,40 ± 2,11 и 0,39 ± 2,40 в групите с ustekinumab 45 mg (стр
Физическа функция и свързано със здравето качество на живот
Пациентите, лекувани с устекинумаб, показват значително подобрение на физическата функция, оценено от индекса на уврежданията на въпросника за здравна оценка (HAQ-DI) на 24-та седмица. е значително по-голям в групата с ustekinumab, отколкото в плацебо групата.Подобрението на HAQ-DI резултата от изходното ниво се поддържа през 52 и 100 седмици.
C "е значително подобрение в DLQI резултата в групата с ustekinumab в сравнение с плацебо на 24 -та седмица, което се поддържа през 52 и 100 седмици. В PsA проучване 2 c" е значително подобрение във функционалната оценка на хроничния резултат. Терапия на болестта - Умора (FACIT-F) в групата на ustekinumab в сравнение с групата на плацебо на 24-та седмица. Процентът на пациентите, които са постигнали значително подобрение на умората (4 точки при FACIT-F), също е значително по-голям в групата на ustekinumab в сравнение с плацебо. Подобренията в оценката FACIT се запазиха до 52 -та седмица.
Педиатрична популация
Европейската агенция по лекарствата е отложила задължението да представи резултатите от проучванията с устекинумаб в една или повече подгрупи от педиатричната популация на възраст 6-11 години за умерен до тежък плакатен псориазис и ювенилен идиопатичен артрит (вж. Точка 4.2 за информация относно педиатричната употреба) .
Плакатен псориазис при педиатрични пациенти
Доказано е, че устекинумаб подобрява свързаните със здравето признаци и симптоми и качеството на живот при педиатрични пациенти на възраст над 12 години с псориазис на плаки.
Ефикасността на устекинумаб е проучена при 110 педиатрични пациенти на възраст от 12 до 17 години с умерен до тежък плакатен псориазис във Фаза 3, многоцентрово, рандомизирано, двойно-сляпо, плацебо-контролирано проучване (CADMUS). Пациентите са рандомизирани да приемат плацебо (n = 37), или препоръчителната доза устекинумаб (вж. Точка 4.2; n = 36), или половината от препоръчителната доза устекинумаб (n = 37) чрез подкожно инжектиране на седмици 0 и 4 и след това на всеки 12 седмици (q12w) На 12 -та седмица, плацебо -лекуваните пациенти са преминали на лечение с устекинумаб.
Пациентите с PASI ≥ 12, PGA ≥ 3 и участие на BSA от поне 10%, които са били кандидати за системна терапия или фототерапия, са били допустими за проучването. Приблизително 60% от пациентите са били изложени на конвенционална системна терапия или фототерапия. Приблизително 11% от пациентите са били изложени на биологични лекарства преди това.
Първичната крайна точка е делът на пациентите, постигнали PGA индекс на 12 -та седмица изчистен или минимален . Вторичните крайни точки включват PASI 75, PASI 90, промяна от изходното ниво в Индекс на качеството на живот на детската дерматология (CDLQI), промяна от изходното ниво в общия резултат на PedsQL (Педиатричен инвентар за качеството на живот) на 12-та седмица. На 12-та седмица субектите, лекувани с устекинумаб, показват значително по-добро подобрение на псориазиса и свързаното със здравето качество на живот, отколкото тези, лекувани с плацебо (Таблица 7).
Всички пациенти бяха проследени за ефикасност до 52 седмици след първото приложение на изследваното средство. Процентът на пациентите с PGA резултат изчистен или минимален и делът на пациентите, които са постигнали PASI 75, показва пропаст между групите с устекинумаб и плацебо при първото посещение след изходно ниво на 4-та седмица, достигайки своя връх на 12-та седмица. Подобренията в PGA, PASI, CDLQI и PedsQL се запазват на 52-та седмица ( Таблица 7).
Таблица 7: Обобщение на първичните и вторичните крайни точки през седмица 12 и седмица 52
на стр
b CDLQI: CDLQI е дерматологичен инструмент за оценка на ефекта на кожен проблем върху здравословното качество на живот в педиатричната популация. CDLQI от 0 или 1 не показва ефект върху качеството на живот на детето.
c p = 0,002
d PedsQL: PedsQL е обща мярка за качеството на живот, свързана със здравето, разработена за употреба при деца и юноши.
и р = 0,028
По време на плацебо-контролиран период до седмица 12, ефикасността и в двете групи при препоръчителната доза и половината от препоръчителната доза като цяло е сравнима по отношение на първичната крайна точка (съответно 69,4% и 67,6%), въпреки че има данни за доза -свързан отговор за критерии за ефикасност на по -високо ниво (например PGA изчистен , PASI 90). След 12 -та седмица ефикасността като цяло е била по -висока и се поддържа по -добре в лечебната група, която получава пълната препоръчителна доза, отколкото в групата, получаваща половината, в която скромната загуба на ефикасност, наблюдавана в края на лечението, е била по -честа, всеки интервал на дозиране от 12 седмици. Профилът на безопасност на препоръчителната доза и половината от препоръчителната доза е сравним.
болест на Крон
Безопасността и ефикасността на устекинумаб бяха оценени в три многоцентрови, рандомизирани, двойно-слепи, плацебо-контролирани проучвания при възрастни пациенти с умерена до тежка активна болест на Crohn (Индекс на активност на болестта на Crohn [CDAI] = индекс на активност на болестта на Crohn ≥ 220 и ≤ 450 ). Програмата за клинично развитие се състои от две 8-седмични интравенозни индукционни проучвания (UNITED-1 и UNITED-2), последвани от 44-седмично рандомизирано подкожно поддържащо проучване (IM-UNITED), състоящо се от 52 седмици терапия. Индукционните проучвания включват 1409 пациенти (UNITED-1, n = 769; UNITED-2 n = 640). Първичната крайна точка и в двете индукционни проучвания е делът на субектите в клиничен отговор (дефиниран като намаляване на CDAI с ≥ 100 точки) на седмица 6. Данните за ефикасността бяха събрани и анализирани до 8 седмица и за двете проучвания. Допускат се едновременни дози перорални кортикостероиди, имуномодулатори, аминосалицилати и антибиотици и 75% от пациентите продължават да получават поне едно от тези лекарства. И в двете проучвания пациентите бяха рандомизирани да получават единична интравенозна доза от варираща в зависимост от теглото препоръчителна доза от приблизително 6 mg / kg (вж. Точка 4.2 от SmPC на концентрат STELARA 130 mg за инфузионен разтвор) или фиксирана доза от 130 mg устекинумаб или плацебо на седмица 0.
Пациентите на UNITED-1 не са повлияли или са имали непоносимост към предишна анти-TNFα терапия. Приблизително 48% от пациентите не са отговорили на предишна терапия с едно анти-TNFα, а 52% не са отговорили на предишни терапии с 2 или 3 анти-TNF-α. В това проучване 29,1% от пациентите са имали неадекватен първоначален отговор (първични неотговорили), 69,4% са отговорили, но са „загубили отговор“ (вторични, които не са отговорили), а 36,4% са били непоносими към терапиите с анти-TNFa.
Пациентите на UNITED-2 са се провалили поне с една конвенционална терапия, включително кортикостероиди или имуномодулатори, и са били или анти-TNF-α наивни (68,6%), или са получавали преди това, но не неуспешно, анти-TNFα терапия. (31,4%).
Както при UNITED-1, така и при UNITED-2, значително по-висок дял от пациентите са имали клиничен отговор и ремисия в групата на ustekinumab в сравнение с плацебо (Таблица 8). Клиничните отговори и ремисии бяха значими още на 3-та седмица при пациенти, лекувани с устекинумаб, и продължиха да се подобряват до 8-та седмица. В тези индукционни проучвания ефикасността беше по-голяма и по-добре поддържана в групата с променлива доза, отколкото в групата с доза от 130 mg и Поради това се препоръчва променлива доза за интравенозно въвеждане.
Таблица 8: Индуциране на клиничен отговор и ремисия в UNITED-1 и UNITED-2
Клиничната ремисия се определя като индекс CDAI
Отговор 70 точки се определя като намаляване на индекса CDAI с поне 70 точки
* неуспехи срещу TNFα
** неуспехи на конвенционалната терапия
на стр
b стр
Проучването за поддържане (IM-UNITED) оценява 388 пациенти, които постигат клиничен отговор от 100 точки на седмица 8 от индукцията на устекинумаб в проучванията UNITED-1 и UNITED-2. Пациентите са рандомизирани на подкожна поддържаща схема от 90 mg устекинумаб на всеки 8 седмици или 90 mg устекинумаб на всеки 12 седмици или плацебо в продължение на 44 седмици (за препоръчителната поддържаща доза, вижте точка 4.2). По -висок процент от пациентите поддържат клинична ремисия и клиничен отговор в групите с устекинумаб в сравнение с групата на плацебо на седмица 44 (вж. Таблица 9).
Таблица 9: Поддържане на клиничния отговор и ремисия в IM-Uniti (седмица 44; 52 седмици от началото на индукционната доза)
Клиничната ремисия се определя като индекс CDAI
* Плацебо групата се състои от пациенти, които са били в отговор на устекинумаб и са рандомизирани да получават плацебо в началото на поддържащата терапия.
† Пациенти, които са имали клиничен отговор на 100 точки устекинумаб в началото на поддържащата терапия
‡ Пациенти, които са се провалили с конвенционалната терапия, но не и с анти-TNF α терапията
§ Пациенти, които са рефрактерни / непоносими към анти-TNF α
на стр
b стр
c номинално значими (стр
В IM-UNITED 29 от 129 пациенти не поддържат отговор на устекинумаб при лечение на всеки 12 седмици и им е позволено да коригират дозата, за да получават устекинумаб на всеки 8 седмици.
Загубата на отговор се определя като CDAI ≥ 220 точки и повишение на CDAI ≥ 100 пункта от изходното ниво.При тези пациенти клинична ремисия е постигната при 41.4% от пациентите 16 седмици след лечението, коригиране на дозата.
Пациенти, които не са имали клиничен отговор след индукция на устекинумаб на 8-та седмица в индукционните проучвания на UNITED-1 и UNITED-2 (476 пациенти), са влезли в нерандомизираната част от поддържащото проучване (IM-UNITED) и след това са получили подкожна инжекция от 90 mg устекинумаб. Осем седмици по -късно 50,5% от пациентите постигат клиничен отговор и продължават да получават поддържащата доза на всеки 8 седмици; сред тези пациенти на продължителна поддържаща доза, по -голямата част поддържат отговор (68,1%) и постигат ремисия (50,2%) на 44 -та седмица, при нива, които са подобни на пациентите, които първоначално са отговорили на индукция с устекинумаб.
От 131 пациенти, които са отговорили на устекинумаб във фазата на индукция и които са били рандомизирани в плацебо групата в началото на поддържащото проучване, 51 впоследствие не са отговорили и са получавали устекинумаб 90 mg подкожно на всеки 8 седмици. Някои от пациентите, които са загубили отговор и възобновяване на устекинумаб направи това в рамките на 24 седмици след индукционната инфузия. От тези 51 пациенти 70,6% са постигнали клиничен отговор и 39,2% са постигнали клинична ремисия 16 седмици след получаване на първата подкожна доза устекинумаб.
Ендоскопия
Ендоскопският външен вид на лигавицата е оценен в подпроучване при 252 допустими пациенти с изходна активност на ендоскопска болест. 5-те сегмента на илеоколики с наличие / размер на язви, процент от повърхността на лигавицата, покрита с язви, процент от повърхността на лигавицата, засегната от всякакви други лезии и наличие / вид стесняване / стеноза. На седмица 8, след еднократна интравенозна индукционна доза, промяната в SES -CD индекса е по -голяма в групата с устекинумаб (n = 155, средна промяна = -2,8), отколкото в плацебо групата (n = 97, средна промяна = -0,7 , р = 0,012).
Отговор при фистулизираща болест
При подгрупа пациенти с дрениращи фистули в началото (8,8%; n = 26), 12/15 (80%) от лекуваните с устекинумаб пациенти са постигнали отговор след 44 седмици (дефинирано като ≥ 50%намаление от изходното в индукционното проучване в броя на дрениращите фистули) в сравнение с 5/11 (45,5%), изложени на плацебо.
Качеството на живот, свързано със здравето
Качеството на живот, свързано със здравето, беше оценено с помощта на въпросници IBDQ и SF-36. На 8-та седмица пациентите, лекувани с устекинумаб, показват статистически значими по-големи клинични подобрения в общия индекс на IBDQ и в обобщената оценка на умствените компоненти на SF-36 както в UNITED-1, така и в UNITED-2 и SF-36 в резюмето на физическия компонент в UNITED -2, в сравнение с плацебо Тези подобрения обикновено се поддържат по-добре при пациенти, лекувани с устекинумаб в проучването IM-Uniti до 44-та седмица в сравнение с плацебо.
Педиатрична популация
Европейската агенция по лекарствата отложи задължението да представи резултатите от проучванията с устекинумаб в една или повече подгрупи от педиатричната популация при болестта на Crohn (вж. Точка 4.2 за информация относно педиатричната употреба).
05.2 "Фармакокинетични свойства -
Абсорбция
При здрави индивиди средното време за достигане на максимална серумна концентрация (Tmax) е 8,5 дни след еднократно подкожно приложение от 90 mg. Средните Т стойности на устекинумаб след еднократно подкожно приложение на 45 mg или 90 mg при пациенти с псориазис са сравними с тези, наблюдавани при здрави индивиди.
Абсолютната бионаличност на устекинумаб при пациенти с псориазис след еднократно подкожно приложение се оценява на 57,2%.
Разпределение
Средният обем на разпределение по време на терминалната фаза (Vz) след еднократно интравенозно приложение при пациенти с псориазис варира от 57 до 83 ml / kg.
Биотрансформация
Точният метаболитен процес на устекинумаб не е известен.
Елиминиране
Там освобождаване Средната системна (CL) при пациенти с псориазис след еднократно интравенозно приложение варира от 1,99 до 2,34 ml /умират/ килограма.
Средният полуживот (t1 / 2) на устекинумаб е приблизително 3 седмици при пациенти с псориазис, псориатичен артрит или болест на Crohn, вариращ от 15 до 32 дни във всички проучвания за псориазис и псориатичен артрит.
В „анализ на популационния фармакокинетичен профил при пациенти с псориазис, освобождаване видимият (CL / F) и видимият обем на разпределение (V / F) са съответно 0,465 L / ден и 15,7 L. CL / F на ustekinumab не е повлиян от пола. Популационният фармакокинетичен анализ показа тенденция към увеличаване на клирънса на устекинумаб при пациенти, позитивни на антиустекинумаб антитела.
Линейност на дозата
Системната експозиция на устекинумаб (Cmax и AUC) нараства сравнително дозата пропорционално след еднократно интравенозно приложение на дози, вариращи от 0.09 mg / kg до 4.5 mg / kg или след еднократно приложение подкожно в дози, вариращи от около 24 mg до 240 mg при пациенти с псориазис.
Единична доза спрямо многократни дози
Профилите на серумната концентрация на устекинумаб във времето са широко предсказуеми след еднократни или многократни подкожни дози. При пациенти с псориазис серумни концентрации в стационарно състояние (стабилно състояние) на устекинумаб бяха постигнати, започвайки от 28 -та седмица след подкожно дозиране на 0 и 4 седмици, последвано от дозиране на всеки 12 седмици. Минималната средна концентрация в стационарно състояние (стабилно състояние) е между 0.21 mcg / mL и 0.26 mcg / mL (45 mg) и между 0.47 mcg / mL и 0.49 mcg / mL (90 mg).
След подкожно приложение на всеки 12 седмици не се наблюдава видимо натрупване на серумна концентрация на устекинумаб във времето. При пациенти с болест на Crohn, след интравенозна доза ~ 6 mg / kg, поддържаща доза от 90 mg устекинумаб се прилага подкожно на всеки 8 или 12 седмици, започвайки от 8 седмица. Стационарната концентрация (стабилно състояние) на устекинумаб е достигнат в началото на втората поддържаща доза.стабилно състояние) на устекинумаб варира от 1,97 mg / mL до 2,24 mg / mL и от 0,61 mg / mL до 0,76 mg / mL за 90 mg устекинумаб съответно на всеки 8 седмици или на всеки 12 седмици. Стационарно най-ниско ниво на устекинумаб (стабилно състояние) резултатите от ustekinumab 90 mg на всеки 8 седмици са свързани с по-високи нива на клинична ремисия от минималните нива на равновесно състояние от 90 mg на всеки 12 седмици.
Влияние на теглото върху фармакокинетичния профил
В „ПК анализ на популацията на пациентите, използващ данни от пациенти с псориазис, беше установено, че телесното тегло е ковариантното, което най -значително повлиява освобождаване от ustekinumab. Средната CL / F при пациенти с тегло> 100 kg е приблизително 55% по -висока от тази при пациенти с тегло ≤ 100 kg. Средната V / F при пациенти с тегло> 100 kg е приблизително 37% по -висока от тази при пациенти с тегло ≤ 100 kg. По -ниските средни серумни концентрации на устекинумаб при пациенти с по -голямо тегло (> 100 kg) в групата с 90 mg дози са сравними с тези при пациенти с по -ниско тегло (≤ 100 kg) в лекуваната група. С доза от 45 mg. Подобни резултати са получени от потвърждаващ популационен фармакокинетичен анализ, използващ данни от пациенти с псориатичен артрит.
Специални популации
Няма налични фармакокинетични данни при пациенти с бъбречна или чернодробна дисфункция.
Не са провеждани специфични клинични проучвания при пациенти в напреднала възраст.
Фармакокинетичният профил на устекинумаб като цяло е сравним между азиатските и неазиатските пациенти с псориазис.
При пациенти с болест на Crohn, променливостта на CL на устекинумаб е повлияна от телесното тегло, нивото на серумния албумин, CRP, предишната недостатъчност на антагонистите на TNF, пола, расата (азиатски срещу неазиатски) и наличието на антитела към устекинумаб, докато телесното тегло е основното едновременното използване на имуномодулатори не оказва значително влияние върху разпределението на устекинумаб. Въздействието на тези статистически значими ковариати върху съответните им фармакокинетични параметри е в рамките на ± 20%, когато се оценяват в представителен диапазон от данни за ковариати или категории, който е в общата наблюдавана вариабилност в ПК на устекинумаб. При фармакокинетичния анализ на популацията пациенти не са наблюдавани индикации за ефект на тютюн или алкохол върху фармакокинетичния профил на устекинумаб.
Серумните концентрации на устекинумаб при педиатрични пациенти на възраст от 12 до 17 години с псориазис, лекувани с препоръчителната доза въз основа на телесното тегло, като цяло са сравними с тези в възрастната популация от псориазис, лекувани с препоръчителната доза за възрастни, докато серумните концентрации на устекинумаб при деца пациентите с псориазис, лекувани с половината от препоръчителната доза въз основа на телесното тегло, обикновено са по -ниски, отколкото при възрастни.
Регулиране на ензимите CYP450
Ефектите на IL-12 или IL-23 върху регулацията на ензимите CYP450 бяха оценени в едно проучване инвитро използвайки човешки хепатоцити, това проучване показа, че IL-12 и / или IL-23 при нива от 10 ng / mL не променят ензимната активност на човешки CYP450 (CYP1A2, 2B6, 2C9, 2C19, 2D6 или 3A4; вижте точка 4.5 ).
05.3 Предклинични данни за безопасност -
Неклиничните данни не разкриват особен риск за хората (например токсичност за органи) въз основа на токсичност при многократни дози и проучвания за токсичност за развитието и репродуктивната функция, включително оценки на фармакология за безопасност. При проучвания за репродуктивна токсичност и развитие, проведени при маймуни cynomolgus, не са наблюдавани неблагоприятни ефекти върху индексите на фертилитета при мъжете, вродени дефекти или токсичност за развитието. Не са наблюдавани неблагоприятни ефекти върху женските индекси на фертилитета при използване на антитяло, аналогично на IL-12/23 при мишки.
Нивата на дозата в проучвания при животни са били приблизително 45 пъти по -високи от най -високата еквивалентна доза, предназначена за пациенти с псориазис. При маймуните тези нива се трансформират в пикови серумни концентрации, които са 100 пъти или повече по -високи от тези, наблюдавани при хора.
Не са провеждани проучвания за канцерогенност на устекинумаб поради липсата на подходящи модели на антитела, свободни от кръстосано реагиращ IL-12/23 р40 при гризачи.
06.0 ФАРМАЦЕВТИЧНА ИНФОРМАЦИЯ -
06.1 Помощни вещества -
L-хистидин
L-хистидин монохидрохлорид монохидрат
Полисорбат 80
Захароза
Вода за инжекции
06.2 Несъвместимост "-
При липса на проучвания за съвместимост, този лекарствен продукт не трябва да се смесва с други лекарствени продукти.
06.3 Срок на валидност "-
2 години
06.4 Специални условия на съхранение -
Съхранявайте в хладилник (2 ° C - 8 ° C). Не замразявайте.
Съхранявайте флакона или предварително напълнената спринцовка във външната опаковка, за да предпазите лекарството от светлина.
06.5 Естество на непосредствената опаковка и съдържанието на опаковката -
STELARA 45 mg инжекционен разтвор
0,5 ml разтвор във флакон от 2 ml, изработен от стъкло тип I, затворен с запушалка от бутилова гума.
STELARA 90 mg инжекционен разтвор
1 ml разтвор във флакон от 2 ml, изработен от стъкло тип I, затворен с запушалка от бутилова гума.
STELARA 45 mg инжекционен разтвор в предварително напълнена спринцовка
0,5 ml разтвор в 1 ml стъклена спринцовка тип I, с неразглобяема стоманена игла, защитена с капачка, съдържаща изсушен естествен каучук (производно на латекс). Спринцовката е оборудвана с пасивно предпазно устройство.
STELARA 90 mg инжекционен разтвор в предварително напълнена спринцовка
1 mL разтвор в 1 mL стъклена спринцовка тип I с неразглобяема стоманена игла, защитена с капачка, съдържаща изсушен естествен каучук (производно на латекс). Спринцовката е оборудвана с пасивно предпазно устройство.
STELARA се предлага в опаковки от 1 флакон или 1 предварително напълнена спринцовка.
06.6 Инструкции за употреба и боравене -
Разтворът, съдържащ се във флакона STELARA или предварително напълнената спринцовка, не трябва да се разклаща. Разтворът трябва да бъде визуално проверен за наличие на частици или обезцветяване преди подкожно приложение. Разтворът е бистър до леко опалесциращ, безцветен до бледожълт и може да съдържа някои малки полупрозрачни или бели протеинови частици. Не е необичайно за протеиновите разтвори. продуктът не трябва да се използва, ако разтворът е обезцветен или непрозрачен, или ако има чужди прахови частици.Преди приложение STELARA трябва да се остави да достигне стайна температура (приблизително половин час). Подробни инструкции за употреба са дадени в листовката.
STELARA не съдържа консерванти, така че неизползваното лекарство, останало във флакона и спринцовката, не трябва да се използва. STELARA се доставя като стерилен флакон за еднократна употреба или предварително напълнена спринцовка за еднократна употреба. Спринцовката, иглата и флаконът никога не трябва да се използват повторно. Неизползваното лекарство и отпадъците от това лекарство трябва да се изхвърлят в съответствие с местните изисквания.
07.0 ПРИТЕЖАТЕЛ НА „РАЗРЕШЕНИЕТО ЗА ТЪРГОВЕ“
Janssen-Cilag International NV
Turnhoutseweg 30
2340 Бира
Белгия
08.0 НОМЕР НА РАЗРЕШЕНИЕ ЗА ТЪРГОВЕ -
STELARA 45 mg инжекционен разтвор
EU/1/08/494/001
STELARA 90 mg инжекционен разтвор
EU/1/08/494/002
STELARA 45 mg инжекционен разтвор в предварително напълнена спринцовка
EU/1/08/494/003
STELARA 90 mg инжекционен разтвор в предварително напълнена спринцовка
EU/1/08/494/004
038936035
038936047
038936011
038936023
09.0 ДАТА НА ПЪРВО РАЗРЕШЕНИЕ ИЛИ ПОДНОВЯВАНЕ НА РАЗРЕШЕНИЕТО -
Дата на първо разрешаване: 16 януари 2009 г.
Дата на последното подновяване: 19 септември 2013 г.